We offer a range of services such as sample preparation, mass spectrometry measurement as well as data analysis and presentation. Our methods can be applied to a wide variety of samples including purified proteins and protein complexes, cells, tissues and biofluids.
1) Identification of proteins and their post-translational modifications (PTMs)
We offer mass spectrometry-based identification of proteins and PTMs within a given sample, including acetylation, glycosylation, phosphorylation, proteolytic cleavage, and ubiquitination. Only high confidence results are reported using state-of-the-art data analysis tools and strategies.
1. Global analysis of the mitochondrial N-proteome identifies a processing peptidase critical for protein stability. Vögtle, F.-N., Wortelkamp, S., Zahedi, R.P., Becker, D., Leidhold, C., Gevaert, K., Kellermann, J., Voos, W., Sickmann, A., Pfanner, N., Meisinger, C., 2009. Cell 139, 428–439.
2. Simple, scalable, and ultrasensitive tip-based identification of protease substrates. Shema, G., Nguyen, M.T.N., Solari, F.A., Loroch, S., Venne, A.S., Kollipara, L., Sickmann, A., Verhelst, S.H.L., Zahedi, R.P., 2018. Mol. Cell. Proteomics MCP 17, 826–834.
3. PARL mediates Smac proteolytic maturation in mitochondria to promote apoptosis. Saita, S., Nolte, H., Fiedler, K.U., Kashkar, H., Venne, A.S., Zahedi, R.P., Krüger, M., Langer, T., 2017. Nat. Cell Biol. 19, 318–328.
4. Effective Assignment of α2,3/α2,6-Sialic Acid Isomers by LC-MS/MS-Based Glycoproteomics. Pett, C., Nasir, W., Sihlbom, C., Olsson, B.-M., Caixeta, V., Schorlemer, M., Zahedi, R.P., Larson, G., Nilsson, J., Westerlind, U., 2018. Angew. Chem. Int. Ed Engl. 57, 9320–9324.
5. Enhanced N-glycosylation site analysis of sialoglycopeptides by strong cation exchange prefractionation applied to platelet plasma membranes. Lewandrowski, U., Zahedi, R.P., Moebius, J., Walter, U., Sickmann, A., 2007. Mol. Cell. Proteomics MCP 6, 1933–1941.
6. Regulation of mitochondrial protein import by cytosolic kinases. Schmidt, O., Harbauer, A.B., Rao, S., Eyrich, B., Zahedi, R.P., Stojanovski, D., Schönfisch, B., Guiard, B., Sickmann, A., Pfanner, N., Meisinger, C., 2011. Cell 144, 227–239.
2) Relative quantitative proteomics and phosphoproteomics for discovery
In-depth analysis of cells, tissues and biofluids will lead to the identification of regulated proteins and pathways. Depending on sample type, up to 10,000 proteins and more than 10,000 phosphorylation sites can be quantified from as little as 25 µg (proteome) to 100 µg (phosphoproteome) of total protein per lysate. Only high confidence identifications using state-of-the-art data analysis tools and strategies are reported. Significantly regulated proteins and phosphorylation sites will be reported using fold-change cutoffs based on the data distribution and statistical tests. Visualization of regulated pathways etc. is available on request.
Tracking Effects of SIL1 Increase: Taking a Closer Look Beyond the Consequences of Elevated Expression Level. Labisch, T., Buchkremer, S., Phan, V., Kollipara, L., Gatz, C., Lentz, C., Nolte, K., Vervoorts, J., Coraspe, J.A.G., Sickmann, A., Carr, S., Zahedi, R.P., Weis, J., Roos, A., 2018. Mol Neurobiol. 55(3), 2524-2546.
1. Quantifying Missing (Phospho)Proteome Regions with the Broad-Specificity Protease Subtilisin. Gonczarowska-Jorge, H., Loroch, S., Dell’Aica, M., Sickmann, A., Roos, A., Zahedi, R.P., 2017. Anal. Chem. 89, 13137–13145.
2. Time-resolved characterization of cAMP/PKA-dependent signaling reveals that platelet inhibition is a concerted process involving multiple signaling pathways. Beck, F., Geiger, J., Gambaryan, S., Veit, J., Vaudel, M., Nollau, P., Kohlbacher, O., Martens, L., Walter, U., Sickmann, A., Zahedi, R.P., 2014. Blood 123, e1–e10.
3. Temporal quantitative phosphoproteomics of ADP stimulation reveals novel central nodes in platelet activation and inhibition. Beck, F., Geiger, J., Gambaryan, S., Solari, F.A., Dell’Aica, M., Loroch, S., Mattheij, N.J., Mindukshev, I., Pötz, O., Jurk, K., Burkhart, J.M., Fufezan, C., Heemskerk, J.W.M., Walter, U., Zahedi, R.P., Sickmann, A., 2017. Blood 129, e1–e12.
3) Targeted quantitative mass spectrometry
We have developed a large number of assays for precise absolute quantification of selected proteins using stable isotope-labeled reference standard(SIS) peptides in combination with targeted mass spectrometry (Multiple Reaction Monitoring, MRM; Parallel Reaction Monitoring, PRM).
These highly-specific assays allow the precise and sensitive determination of protein concentrations in mole per volume/cell/weight, down to the attomolar range. MRM and PRM can be used for high-throughput (multiplexed) quantification of proteins without the need for antibodies. Moreover, they are more precise and specific than Western blots. Targeted MS using SIS peptides allows the determination of phosphorylation stoichiometries.
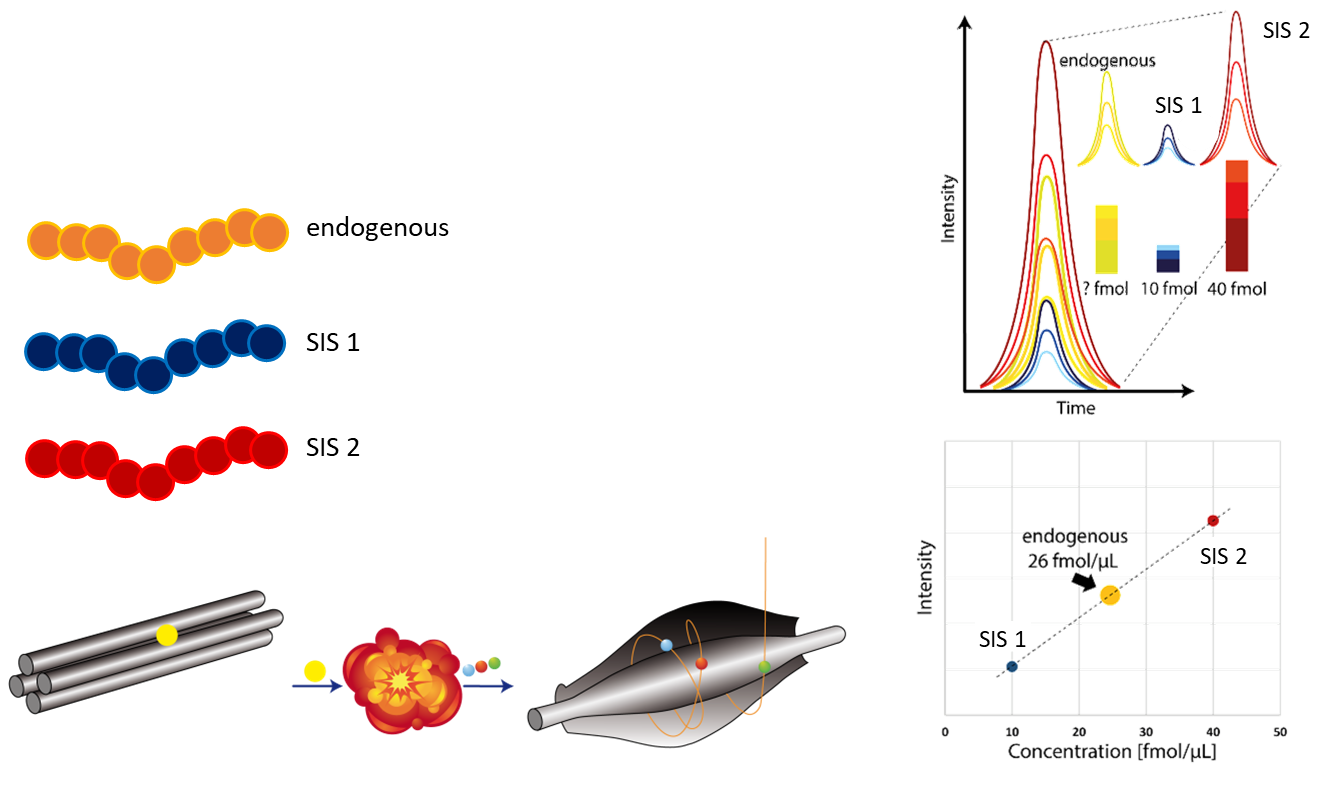
1. The mTOR and PP2A Pathways Regulate PHD2 Phosphorylation to Fine-Tune HIF1α Levels and Colorectal Cancer Cell Survival under Hypoxia. Di Conza, G., Trusso Cafarello, S., Loroch, S., Mennerich, D., Deschoemaeker, S., Di Matteo, M., Ehling, M., Gevaert, K., Prenen, H., Zahedi, R.P., Sickmann, A., Kietzmann, T., Moretti, F., Mazzone, M., 2017. Cell Rep. 18, 1699–1712.
2. Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Kuzyk, M.A., Smith, D., Yang, J., Cross, T.J., Jackson, A.M., Hardie, D.B., Anderson, N.L., Borchers, C.H., 2009. Mol. Cell. Proteomics MCP 8, 1860–1877.
3. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Addona, T.A., Abbatiello, S.E., Schilling, B., Skates, S.J., Mani, D.R., Bunk, D.M., Spiegelman, C.H., Zimmerman, L.J., Ham, A.-J.L., Keshishian, H., Hall, S.C., Allen, S., Blackman, R.K., Borchers, C.H., Buck, C., Cardasis, H.L., Cusack, M.P., Dodder, N.G., Gibson, B.W., Held, J.M., Hiltke, T., Jackson, A., Johansen, E.B., Kinsinger, C.R., Li, J., Mesri, M., Neubert, T.A., Niles, R.K., Pulsipher, T.C., Ransohoff, D., Rodriguez, H., Rudnick, P.A., Smith, D., Tabb, D.L., Tegeler, T.J., Variyath, A.M., Vega-Montoto, L.J., Wahlander, A., Waldemarson, S., Wang, M., Whiteaker, J.R., Zhao, L., Anderson, N.L., Fisher, S.J., Liebler, D.C., Paulovich, A.G., Regnier, F.E., Tempst, P., Carr, S.A., 2009. Nat. Biotechnol. 27, 633–641.
4. MRM-based multiplexed quantitation of 67 putative cardiovascular disease biomarkers in human plasma. Domanski, D., Percy, A.J., Yang, J., Chambers, A.G., Hill, J.S., Freue, G.V.C., Borchers, C.H., 2012. Proteomics 12, 1222–1243.
4) Multi-OMICS
Our MultiOMICS service consists of the combined analysis of the proteome, metabolome and lipidome from a single sample (no replicates, all from one vial) using our SIMPLEX (Simultaneous Metabolite, Protein, Lipid Extraction) strategy. The complementary datasets provide a view of the interconnectivity of different biomolecular classes, which provide an extremely detailed view of the phenotype of the sample.
Simultaneous Metabolite, Protein, Lipid Extraction (SIMPLEX): A Combinatorial Multimolecular Omics Approach for Systems Biology. Coman, C., Solari, F.A., Hentschel, A., Sickmann, A., Zahedi, R.P., Ahrends, R., 2016. Mol. Cell. Proteomics MCP 15, 1453–1466.
5) Protein-protein interaction studies / structural proteomics
We employ chemical cross-linking of proteins to identify interaction partners and protein complex components using mass spectrometry. This approach also allows for prediction of protein structures not easily determined by X-ray crystallography or other “traditional” approaches.
1. Isotopically coded cleavable cross-linker for studying protein-protein interaction and protein complexes. Petrotchenko, E.V., Olkhovik, V.K., Borchers, C.H., 2005. Mol. Cell. Proteomics MCP 4, 1167–1179.
2. Structure of EspB from the ESX-1 type VII secretion system and insights into its export mechanism. Solomonson, M., Setiaputra, D., Makepeace, K.A.T., Lameignere, E., Petrotchenko, E.V., Conrady, D.G., Bergeron, J.R., Vuckovic, M., DiMaio, F., Borchers, C.H., Yip, C.K., Strynadka, N.C.J., 2015. Struct. Lond. Engl. 1993 23, 571–583. https://doi.org/10.1016/j.str.2015.01.002.
3. Super Spy variants implicate flexibility in chaperone action. Quan, S., Wang, L., Petrotchenko, E.V., Makepeace, K.A., Horowitz, S., Yang, J., Zhang, Y., Borchers, C.H., Bardwell, J.C., 2014. eLife 3, e01584.
4. Architecture of the RNA polymerase II-Mediator core initiation complex. Plaschka, C., Larivière, L., Wenzeck, L., Seizl, M., Hemann, M., Tegunov, D., Petrotchenko, E.V., Borchers, C.H., Baumeister, W., Herzog, F., Villa, E., Cramer, P., 2015. Nature 518, 376–380.
6) Immuno-MS assays
Our immuno-MS assays are based on immuno-enrichment of targets using anti-peptide and anti-protein antibodies, combined with mass spectrometry for the highly sensitive and quantification of proteins and their PTMs, from clinically relevant sample types (e.g., needle biopsies). Anti-peptide immuno-MS assays are established for AKT1, AKT2, and under development for P13K, PD-L1 and PTEN (peptide+protein).
1. An immunoaffinity tandem mass spectrometry (iMALDI) assay for detection of Francisella tularensis. Jiang, J., Parker, C.E., Fuller, J.R., Kawula, T.H., Borchers, C.H., 2007. Anal. Chim. Acta 605, 70–79.
2. Towards the development of an immuno MALDI (iMALDI) mass spectrometry assay for the diagnosis of hypertension. Reid, J.D., Holmes, D.T., Mason, D.R., Shah, B., Borchers, C.H., 2010. J. Am. Soc. Mass Spectrom. 21, 1680–1686.
3. Development and evaluation of an immuno-MALDI (iMALDI) assay for angiotensin I and the diagnosis of secondary hypertension. Camenzind, A.G., van der Gugten, J.G., Popp, R., Holmes, D.T., Borchers, C.H., 2013. Clin. Proteomics 10, 20.
4. Immuno-Matrix-Assisted Laser Desorption/Ionization Assays for Quantifying AKT1 and AKT2 in Breast and Colorectal Cancer Cell Lines and Tumors. Popp, R., Li, H., LeBlanc, A., Mohammed, Y., Aguilar-Mahecha, A., Chambers, A.G., Lan, C., Poetz, O., Basik, M., Batist, G., Borchers, C.H., 2017. Anal. Chem. 89, 10592–10600.
7) Metabolomics
As a member of the Metabolomics Innovation Centre (TMIC), we offer quantitative MS-based metabolomics assays for hundreds of small molecules, with a variety of panels representing different metabolic pathways or classes of molecules.